RETÍCULO
ENDOPLÁSMICO
Es una red de membranas que
abarca gran parte del citoplasma. Dentro está un espacio extenso o “luz”,
separado del citosol vecino por la membrana del RE.
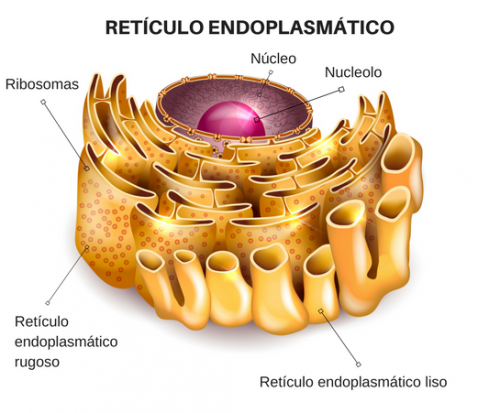
RETÍCULO
ENDOPLÁSMICO LISO
•Carece
de ribosomas
•Elementos membranosos
curvos y tubulares
•Vesículas de superficie
lisa
•Presenta proteínas para la
flexión de la membrana (reticulones)
Donde
lo podemos encontrar
•Músculo esquelético
•Túbulos renales
•Gonadas
Funciones
·
Síntesis de hormonas esteroideas en células
endocrinas de las gónadas y la corteza suprarrenal
·
Desintoxicacion en el hígado de compuestos
orgánicos
•Barbitúricos y etanol
•Oxigenasas, citocromo P-450 que oxidan compuestos hidrófobos y los
convierten en hidrófilos
o
Consecuencias
Pero como no es específico puede actuar con algunos sustratos y
convertirlos en carcinógenos potentes
·
Reticulo
sarcoplasmico células musculares
•Secuestra iones calcio, para después liberarlo produciendo así la
contracción muscular
•Metabolismo de lípidos y esteroides
•Metabolismo de glucógeno
•Formación y reciclaje de membranas
•Asociada con metilasas, hidrolasas, glucosa 6-fosfatasa y oxidasas de
lípidos.
RETÍCULO
ENDOPLÁSMICO RUGOSO (RER)
Funciones
·
Investigaciones realizadas en células que
secretan grandes cantidades de proteínas (Células acinares del páncreas o
células secretoras de moco del tubo digestivo)
·
El retículo endoplásmico rugoso es el punto
inicial de la vía biosintética*
·
Sus membranas son el sitio de producción de
todas las proteínas transmembranales y de secreción.
·
Es el punto donde se sintetizan las
proteínas, cadenas de carbohidratos y fosfolípidos que viajan por los
compartimientos membranosos de la célula.
·
Las proteínas
precursoras son dirigidas desde el citosol a la membrana del RER (targeting) e
insertadas o transportadas a través de la membrana al lumen durante o
inmediatamente después de su síntesis en el proceso conocido como translocación.
·
Sitio
de “control de calidad” en donde las proteínas procesadas erróneamente
son enviadas al citoplasma y degradadas en los proteosomas
SÍNTESIS DE PROTEÍNAS EN RIBOSOMAS
Los polipéptidos se sintetizan en dos puntos distintos dentro de la célula:
La tercera parte de las proteínas codificadas por el genoma de mamíferos
son sintetizadas en los ribosomas unidos a la superficie citosólica de las
membranas RER. (Proteínas que secreta la célula, proteínas integrales y
proteínas solubles).
Otros se sintetizan en ribosomas “libres”, los que no están incluidos en el
RER, y luego se liberan al citosol estas incluyen: Proteínas destinadas a permanecer en el citosol (enzimas
de glucólisis y proteínas de citoesqueleto)
- ● Proteínas periféricas (Espectrinas y Anquirinas)
- ● Proteínas que se transportan al núcleo.
- ● Proteínas que se incorporan a los peroxisomas,cloroplastos o mitocondrias.
¿QUÉ DETERMINA EL SITIO DE LA CÉLULA
EN EL QUE SE SINTETIZA UNA PROTEÍNA?
Demostraron que el sitio de la síntesis de una proteína dependía de la
secuencia de aminoácidos en la porción amino-terminal del polipéptido (primera
parte que surge del ribosoma durante la síntesis de proteínas.
Las proteínas secretoras contienen una secuencia de señal en su
extremo amino que dirige al polipéptido emergente y al ribosoma hacia la
membrana del retículo endoplásmico.
El polipéptido se mueve en dirección al espacio de cisterna del retículo
endoplásmico, un canal acuoso recubierto con proteína en la membrana del
retículo endoplásmico. Se mueve por la membrana conforme se sintetiza (al mismo
tiempo de la traducción). HIPÓTESIS DE LA SEÑAL
Blobel:
Las proteínas tienen incluidos “Códigos de postales”.
TARGETING
Transporte de las proteínas precursoras desde el citosol a la membrana del
RER
Los elementos moleculares requeridos para dirigir una cadena polipeptídica
(unida a un ribosoma) a la membrana del RER son:
La partícula de reconocimiento de la señal (SRP):es un complejo
ribonucleoproteico soluble que comprende seis polipéptidos y un RNA de 300
nucleótidos, dirige proteínas secretoras y de membrana al RE.
El receptor de la partícula de reconocimiento de la señal (SRPR) en
mamíferos sólo se ha encontrado en la membrana del RE y es una proteína
integral de membrana compuesta de las subunidades SRα y SRβ. Ambas subunidades
son GTPasas
La secuencia señal (SS): Localizada en el extremo amino-terminal de
las proteínas nacientes, contiene un núcleo de 8 a 30 aminoácidos hidrofóbicos
y que se encuentra en las proteínas que siguen la ruta secretora.

TRANSLOCACIÓN DE PROTEÍNAS
Constituye el primer paso en el proceso de transporte de las proteínas que
habrán de formar parte de otros organelos.
Se define como el proceso mediante el cual un polipéptido naciente se
transporta a través de la bicapa lipídica hacia el lumen (en el caso de
proteínas secretoras), o se inserta en la membrana (en el caso de proteínas
integrales de membrana), evento que puede ocurrir cotraduccionalmente o postraduccionalmente.

PROCESAMIENTO DE PROTEÍNAS RECIÉN
SINTETIZADAS EN EL RE
Conforme entra a la cisterna del RER, un polipéptido naciente es sujeto de
la actividad de diversas enzimas situadas en la luz del RER.
La porción amino-terminal que contiene el péptido de señal se retira la
mayor parte de los polipéptidos nacientes por acción de la enzima peptidasa
de señal.
Los carbohidratos se agregan a la proteína naciente mediante la enzima oligosacariltransferasa
●
El RER
es una planta procesadora de proteínas.
●
La luz
del RER está empacada con chaperonas* moleculares
●
La luz
del RE contiene varias enzimas procesadoras como:
1) PDI (disulfuro isomerasa de proteína)
●
Las
proteínas entran en la luz del RE con sus residuos cisteína (-SH) pero salen
con muchos de estos residuos unidos entre sí como disulfuros oxidados (-SS-).
Catalizado por la PDI (reordenamiento).
Enlaces disulfuro: Tienen la función de mantener la estabilidad de las
proteínas que se encuentran en la superficie extracelular de la membrana
plasmática.
●
La luz
de las cisternas del RE favorece la modificación, el plegamiento y el ensamble
de proteínas de la célula.
CHAPERONAS: MOLÉCULAS QUE AYUDAN A
LAS PROTEÍNAS A PLEGARSE DE MANERA APROPIADA
1962--F.M.Ritossa--Mosca de la fruta (Drosophila)
●
Respuesta
al choque térmico no era exclusiva de la mosca de la fruta.
¿Cual es la función de estas proteínas de choque térmico? Capacidad de
ensamblarse por sí mismas, pero son incapaces de formar una partícula viral.
Ensamble proteico
●
GroEL y
GroES
●
Rubisco:
proteína grande presente en los cloroplastos que cataliza la reacción en la que
las moléculas de CO2 se unen por enlaces covalentes a moléculas orgánicas. 16
unidades (8g y 8p)
●
Proteína
(cadenas pesadas)--Proteína de unión o BiP
●
1986--Proteína
70 de choque térmico era idéntica a la BiP--(ensamble de anticuerpos)
●
Las
proteínas eran sensibles a la temperatura, un pequeño cambio empezarán a
desplegarse
●
Tal
desdoblamiento expone los residuos hidrófobos que estaban ocultos en el centro
de la proteína
●
Se
desnaturalizan y forman agregados.
●
Las
hsp70 y moléculas relacionadas se llamaron chaperonas moleculares
●
Chaperonas
Hsp60-bacterias y Rubisco.
●
Función
básica:
- Mediar el ensamble
de complejos
- 1989-chaperonas
mitocondriales-auxilian en el plegamiento de cadenas polipeptídicas.
Las chaperonas no transmiten información para el proceso de doblamiento,
sino que impiden que las proteínas se desvien de su vía de plegamiento correcto
y se encuentren en estados mal plegados o agregados.----Anfinsen

BIOSÍNTESIS DE MEMBRANA EN EL RE
●
Las
membranas no surgen de novo, sino que surgen de membranas preexistentes.
●
Las
membranas crecen conforme las proteínas y lípidos recién sintetizados se
insertan en las membranas existentes en el ER.
●
Los
componentes de la membrana pasan del RE a todos los demás compartimientos de la
célula.
●
Cuando
la membrana se mueve, sus proteínas y lípidos se modifican por efecto de las
enzimas que residen en los diversos organelos de la célula.
●
Las
membranas celulares son asimétricas.
SÍNTESIS DE LOS LÍPIDOS DE LA
MEMBRANA
La mayor parte de los lípidos de la membrana se sintetiza por completo
dentro del retículo endoplásmico
EXCEPCIONES
La esfingomielina y los glucolípidos
(RE---Aparato de Golgi)
Lípidos de mitocondrias y
cloroplastos, se sintetizan por enzimas
·
Enzimas participantes: Proteínas integrales,
sus sitios activos dirigidos hacia el citosol.
·
Los fosfolípidos recién producidos se
insertan en la mitad de la bicapa dirigidos hacia el citosol.
·
Algunos migran hacia la hoja contraria por
acción de las enzimas flipasas.
·
Son transportados del RE al Aparato de Golgi
y la membrana plasmática.
Las membranas de los diferentes organelos tienen una composición de lípidos
muy diferente y puede contribuir a varios cambios como:
La mayor parte de los organelos tienen enzimas que modifican los lípidos
que ya están dentro de su membrana y convierten un tipo de fosfolípidos.
Cuando las vesículas se desprenden de un compartimiento, algunos tipos de fosfolípidos
pueden incluirse dentro mientras que otros se dejan atrás.
Las células contienen proteínas de transferencia de lípidos (facilitan el
transporte de lípidos desde el RE a otros organelos sin la participación de las
vesículas de transporte) que pueden unir o transportar a los lípidos a través
del citosol acuoso de un compartimiento a otro.


Las proteínas formadas en ribosomas unidos a membrana
Glucoproteinas
● Componentes integrales membrana
● MEC
Función
· Sitios de unión para interactuar con otras moléculas
· Plegamiento correcto de la proteína
OLIGOSACARIDOS
Consistentes y predecibles
Glucosiltransferasas
Transfieren un monosacárido específico de un azúcar-nucleótido, como
GDP-manosa o UDP-N-acetilglucosamina al extremo en crecimiento.
Depende de la localización espacial de las enzimas en la línea de montaje
N-GLICOSILACIÓN
Si hay ausencia provoca la muerte de los embriones antes de la implantación
La interrupción parcial de esta vía en el RE, causa trastorno hereditario
en cualquier órgano y sistema
ENFERMEDADES CONGÉNITAS DE LA
GLUCOSILACIÓN
La interrupción parcial de esta vía en el RE, causa trastorno hereditario
en cualquier órgano y sistema
Pruebas sanguíneas que detectan CDG1b por la deficiencia de la enz. fosfomanosa isomerasa, que convierte la fructosa 6-P en manosa 6-P
Tratamiento:
Complementos orales de manosa
SISTEMA DE CONTROL

ERAD (Degradación vinculada
al RE)
Del RE van al citosol por
transposición inversa
Las cadenas de
oligosacáridos se retiran y las proteínas mal plegadas se degradan en los
proteosomas
MECANISMOS
QUE ASEGURAN LA DESTRUCCION DE PROTEINAS MAL PLEGADAS
UPR (Respuesta de proteína
no plegada)
Se forman a mayor velocidad de la que pueden transportarse al citoplasma
por lo que se acumulan
Las moléculas BiP funcionan como chaperonas manteniendo activo los
receptores

DEL RE AL APARATO DE GOLGI
Sitios de salida del RER sin ribosomas, se forman las primeras vesículas de
transporte
Ya que se desprenden varias vesículas de transporte se fusionan para
hacerse más grandes (transporte versículo tubulares) y formar túbulos
interconectados (Compartimento intermedio)
CREUTZFELD-JAKOB (CJD)
Gen PRNP
ü Neurodegenerativo
ü Pérdida de la coordinación motora
ü Demencia
·
Hereditaria
·
Adquirida
·
Síndrome
de las vacas locas
ALZHEIMER

ü Confusion
ü Pérdida de la capacidad para razonar
Depósitos fibrilares de péptido amiloide B
AB42 Tiene un plegamiento anormal
·
Duplicado
del gen APP
·
Mutaciones
en PSEN1 y PSEN2 encargadas de codificar secretasa gama
APARATO
DE GOLGI
El aparato de Golgi es un
orgánulo presente en todas las células eucariotas. Pertenece al sistema de
endomembranas.
Es conocido como “aparato
reticular interno”
El Complejo de Golgi está
formado por un conjunto de pilas de sacos aplanados denominados cisternas.
El aparato de Golgi fue
visto por primera vez por LA VALLETE SAIN GEORGE en 1865 al estudiar
espermatocitos
Pero fue el italiano CAMILO GOLGI quien logro describirlo en 1889 como un
aparato reticular interno, descubriéndolo al inventar nuevos procedimientos de
tinción para revelar la organización de las células nerviosas dentro del SNC.
En 1898 aplico una tinción metálica a las células nerviosas del cerebelo y
descubrió una red teñida de oscuro localizada cerca del núcleo celular.
Llamándola APARATO DE GOLGI
La técnica de Schiff-ácido
Periódico marca a los glúcidos de Golgi y esto hace que se puedan ver en el
microscopio óptico.
Su estructura membranosa fue
observada por primera vez al microscopio electrónico por Dalto y Félix en 1954.
Aparato de Golgi fue
identificado con claridad en células sin fijación, preparadas por congelamiento
y fractura.
Morfología
del aparato de Golgi
Localizado cerca del núcleo
celular y en células animales cercano a los centrosomas.
Está formado por diminutos
sáculos envueltos por una membrana, aplanados y apilados a modo de platos.
Cada sáculo se denomina
cisterna.
El número de cisternas
apiladas del AG varía dependiendo del tipo celular.
Se divide en dos caras
funcionales CIS y TRANS.
Está formado por lamelas,
vesículas y vacuolas
Formado por dictiosomas.
En células de plantas y
organismos inferiores, estos apilamientos presentan a menudo 20 o más
cisternas.
Compartimentos
del APARATO DE GOLGI
El AG muestra una
organización polarizada, por lo que presenta dos caras:
Cara cis (generalmente
convexa) está orientada hacia el retículo endoplasmico y recibe las proteínas
de exportación.

Cara trans está orientada
hacia los gránulos secretorios o los centriolos.
Cisternas
de Golgi
Entre ambas regiones cis y
trans existen cisternas centrales o medias que pueden variar en número.
La cisterna más cercana al
retículo endoplasmico es generalmente fenestrada y presenta continuidad con la
red cis-Golgi (RCG).
En la región trans, el AG se
extiende y forma una red de estructuras tubo vesiculares conocida como red
trans-Golgi (RTG).
Las cisternas cis, media y
trans representan una serie de subcompartimentos enriquecidos con enzimas
específicas que llevan a cabo modificaciones postraduccionales a proteínas
recién sintetizadas.
Funciones
En 1923 Nassonov presento la
primera evidencia del papel fundamental del aparato de Golgi en la función
secretora de la porción exocrina del páncreas.
En 1929 Bowen adjudico al
aparato de Golgi una importante participación en la formación del acrosoma del
espermatozoide.
Glucosilacion: La función de
este organelo está relacionada con la adición de moléculas de azúcar a los
péptidos en tránsito para la formación de glicoproteínas.
Podemos decir que el aparato de Golgi es una planta procesadora es por eso la diferencia en su composición de los compartimentos de membrana desde la cara Cis a la Trans.
Podemos decir que el aparato de Golgi es una planta procesadora es por eso la diferencia en su composición de los compartimentos de membrana desde la cara Cis a la Trans.
Formación de la pared
celular en vegetales
Transporte de materiales a
través de vesículas
Dependiendo del estado
fisiológico de la célula, el aparato de Golgi se ve sujeto a cambios de
configuración, tamaño y posición.
El mal funcionamiento, ya
sea en las modificaciones postraduccionales, en la clasificación y/o el
transporte de las glicoproteínas, conduce a la disrupción de la homeostasis
celular.
Movimientos
de los materiales a través del aparato de Golgi
Hace varios años ya se
conocía que diferentes materiales, transitan por los compartimentos del aparato
de Golgi pero se tomaron en cuenta dos nociones que demostraron lo ocurrido:
·
Modelo
de maduración de cisternas
Se presuponía que tales
cisternas de Golgi eran estructuras transitorias. Se suponía que tales
cisternas formaban la cara cis de la pila mediante la fusión de los portadores
membranosos desde el retículo endoplasmico
y el ERGIC y que cada cisterna se movía físicamente desde el extremo cis
al trans de la pila y cambiaba de composición conforme avanzaba.
DE ACUERDO CON EL MODELO,
CADA CISTERNA MADURA A LO LARGO DE LA PILA.
·
Modelo
de transporte vesicular
En este modelo el cargamento
(proteínas secretoras, lisosomicas y de membrana) se lanza a través de la pila
de Golgi, desde la CGN hasta la TGN, en vesículas que se desprenden de un
compartimento de membrana y se fusionan con el compartimento contiguo más
avanzado de la pila.
Glucosilacion
1.
Actúa la enzima Manosidasa quitando varios
residuos de manosa en la región CIS Golgi.
2.
Actúa la enzima N-acetil-glucosamina transferasa I agregando
un residuo de N-acetilglucosamina a uno de los residuos de manosa en la región
CIS Golgi
3.
Actúa la enzima Manosidasa II retirando dos
residuos de manosa en el oligosacárido en la región medial Golgi
4.
Actúa la enzima N-acetil-glucosamina
transferasa II agregando otro residuos
de N-acetilglucosamina a la manosa que quedo libre en la región medial Trans Golgi.
5.
Actúa la enzima fucosil -y -galactosil
-transferasa en la región Trans Golgi agregando residuos de galactosa para
obtener un oligosacárido complejo.
6.
Actúa la enzima Sialil-transferasa agrega un
acido sialico en la región mas Trans de Golgi
La Glucosilacion es la
modificación de los hidratos de carbono unidos a glucoproteinas y
Proteoglucanos sintetizados en el retículo endoplasmico.
En la biosíntesis de
glucoproteinas y glucolipidos en este aparato participan más de 200 enzimas.
Las enzimas llamadas
glucosiltransferasas incorporan residuos glucídicos específicos; las
glucosidasas eliminan residuos glucídicos específicos.
El destino de los
glucoproteinas ( proteínas a las que se le ha añadido una cadena de glúcidos )
es ser secretadas o formar parte de la superficie celular.
Una de la funcion de los
carbohidratos de los glucolipidos es que estan relacionadas con el plegamiento
de proteínas en el retículo endoplasmático rugoso.
La secuencia en que se
transfieren los azucares durante el ensamblaje de un oligosacárido depende de
la secuencia de acción de las glucosiltransferasas que participan en este
proceso.
Todo esto dependerá de la
localización de las enzimas específicas.
Los pasos de la
Glucosilacion en el aparato de Golgi pueden ser muy variados y producen
dominios de carbohidrato con una notable diversidad en la secuencia. Una de las
muchas vías posibles de Glucosilacion
A diferencia de los
oligosacáridos con enlaces N, cuya síntesis comienza en el RE, los unidos con
proteínas mediante enlaces O se articulan por completo dentro del aparato de
Golgi.
·
GLUCOSILACION
ER CON ENLACE N
Después del retiro de los
tres residuos de glucosa,
Glucosidasa 1 quita un
residuo de glucosa en el retículo endoplasmatico
Glucosidasa 2 quita dos
residuos de glucosa.
Manosidasa quita una manosa
del oligosacárido formado
El oligosacárido pasa a la
red Cis Golgi
Transporte
vesicular en el aparato de Golgi
El transporte de
macromoléculas entre compartimentos membranosos es mediado por la formación y
fusión de vesículas.
El transporte de proteínas
es mediado por pequeñas vesículas que geman desde un compartimento de membrana
(donador) que es el que produce las vesículas para fusionarse con un
compartimento (aceptor) que recibe la vesícula y su contenido.
La vía biosintética de una
célula eucariota contiene en una serie de distintos organelo limitados por
membrana que participan en la síntesis y modificación y entrega de proteínas
solubles y membranosas en su destino apropiado en la célula.
Los materiales son
transportados entre compartimentos por vesículas que se desprenden de membranas
donadoras y se fusionan con las membranas receptoras.
Gemación
de vesículas
Un punto muy importante en
el transporte vesicular es la gemación, para que se lleve a cabo la formación
de una vesícula, se requiere de la participación de proteínas de recubrimiento.
Está cubierta es utilizada
como medio mecánico para que las vesículas puedan gemar y dirigirse a su
destino.
Las cubiertas de proteína
tienen por lo menos dos funciones distintas:
Actúan como dispositivo
mecánico que hace que la membrana se curve y forme una vesícula desprendible
Proporciona un mecanismo
para seleccionar los componentes que transporta la vesícula.
Estos componentes incluyen:
Cargamento en proteínas
secretoras, lisosomicas y de membrana que deben
transportarse
La estructura necesaria para
dirigir y conectar la vesícula con la membrana receptora correcta.
La cubierta de la vesícula
está formada por dos capas distintas de proteínas: una jaula externa que forma
el marco de la cubierta y una capa interna de adaptadores que sirven para unir
la cara externa de la bicapa lipídica y el cargamento de vesículas.
Vesículas
cubiertas cop (Coat proteins)
Se han identificado
diferentes clases de vesículas cubiertas; se distinguen por las proteínas que
conforman la cubierta, su apariencia en el microscopio electrónico y su función
en el transito celular.
Las tres vesículas cubiertas
estudiadas son las siguientes:
Vesículas cubiertas con COP
II: Desplazan materiales del retículo endoplasmico “hacia adelante al ERGIC y
al aparato de Golgi.
Vesículas cubiertas con COP
I: Mueven materiales en sentido retrogrado: del ERGIC y la pila de Golgi “hacia
atrás” al ER y de las cisternas Golgi trans “de regreso” a las cisternas Golgi
cis. Subsiste el debate en cuanto a las
actividades adicionales de las vesículas COP I.
Vesículas cubiertas con
Clatrina: Movilizan materiales de la TGN a los endosomas, lisosomas y vacuolas
vegetales. También mueven materiales de la membrana plasmática a los
compartimentos citoplasmáticos a lo largo de la vía endocitica. Además se han
implicado en el tránsito de los endosomas y lisosomas.
Los
tres tipos de vesículas tienen diferentes funciones en el transporte:
Las
cubiertas con COP II median el transporte del RE al REGIC y al
aparto de Golgi, incluso el movimiento de enzimas residentes en el aparato de
Golgi en dirección trans a cis y enzimas residentes del ER del ERGIC y el
aparato de Golgi de regreso al retículo endoplasmico.
Las
cubiertas con COP I regresan las proteínas del ERGIC y aparato
de Golgi al RE
Las cubiertas con Catrina se
encargan del transporte de las TGN a lo endosomas y lisosomas.
CONSERVACION
Y RECUPERACION DE LAS PROTEINAS RESIDENTES DEL RETICULO ENDOPLASMICO
Los estudios sugieren que
las proteínas se mantienen en el orgánulo por una combinación de 2 mecanismos:
RETENCION DE MOLECULAS
RESIDENTES QUE SE EXCLUYEN DE LAS VESICULAS DE TRANSPORTE La retención puede
basarse sobre todo en las propiedades físicas de la proteína.
RECUPERACION DE LAS
MOLECULAS PROFUGAS Para devolverlas al compartimento en que se encuentran
normalmente.
Las proteínas solubles
residentes de la luz del ER como la disulfuro isomerasa de proteínas casi
siempre tienen la señal de recuperación KDEL. (Lys-asp-glu-leu).
Las secuencias más
frecuentes de recuperación para las proteínas de la membrana de ER incluyen dos
residuos básicos estrechamente vinculados KKXX (donde k es lisina y X es
cualquier aminoácido).
Cada
compartimento de la membrana en la vía biosintética puede tener sus propias
señales de recuperación lo que ayuda a explicar cómo cada compartimento
mantiene su complemento único de proteínas a pesar del movimiento constante de
vesículas que entran y salen del mismo.
Ordenamiento de proteínas en
la red trans de Golgi (TGN).
Esta red es la última
estación del aparato de Golgi, funciona como una instancia clasificadora y
dirige las proteínas hacia diversos destinos.
La más conocida de las vías
posteriores del aparato de Golgi es la que lleva enzimas lisosomicas.
Ordenamiento
de proteínas en la red trans de Golgi (TGN).
Esta red es la ultima
estación del aparato de Golgi, funciona como una instancia clasificadora y
dirige las proteínas hacia diversos destinos.
La mas conocida de las vías
posteriores del aparato de Golgi es la que lleva enzimas lisosómicas.
Dirección
de las enzimas lisosómicas a los lisosomas.
1.
Los residuos de manosa de la enzima
lisosomica se fosforilan en las cisternas de Golgi.
2.
Los residuos de manosa luego se incorporan en
forma selectiva en la vesícula cubierta con clatrina en la TGN.
3.
Se cree que los receptores para la manosa
6-fosfato tienen doble función.
4.
Los receptores de la manosa 6-fosfato
interactúan de manera especifica con las enzimas lisosomicas en el lado luminal
de la vesícula e interactúan de manera especifica con los adaptadores en la
superficie citosólica de la vesícula. Los receptores de la manosa 6-fosfato se
separan de las enzimas.
5.
Una vez que se separan los receptores para
manosa-6 fosfato de las enzimas estos regresan al aparato de Golgi.
6.
Las enzimas lisosomicas se vacían en un
endosoma y al final en un lisosoma.
7.
Los receptores para manosa 6-fosfato también
están presentes en la membrana plasmática donde pueden capturar enzimas
lisosomicas que se secretan hacia el espacio extracelular y regresan las
enzimas a una vía que las dirige a un lisosoma.
Direccionamiento
de las vesículas a un compartimiento particular.
Movimiento
de la vesícula hacia el compartimiento blanco especifico.
Las vesículas membranosas deben
viajar distancias considerables en el citoplasma antes de llegar a su objetivo
final.
Estos tipos de movimientos
están mediados sobre todo por micro túbulos que actúan como las vías del tren
que llevan contenedores con cargamento a lo largo de un trayecto definido hacia
un destino predeterminado.
Fijación
de las vesículas al compartimiento blanco.
Se cree que los contactos
iniciales entre una vesícula de transporte y sus membranas blancas, con
cisterna de Golgi están mediados por proteínas fijadoras.
Se han descrito grupos de
proteínas fijadoras: proteínas fibrosas cilíndricas, capaces de formar un
puente molecular, ente las dos membranas a una distancia considerable y grandes
complejos de múltiples proteínas que parecen mantener próximas las dos membranas.
Diferentes proteínas
fijadoras inician la fusión entre diversos tipos de membranas.
Acoplamiento
de las vesículas al compartimiento blanco.
En algún momento durante el
proceso que conduce la fusión vesicular, las membranas de la vesícula y el
compartimento blanco entran en contacto estrecho debido a la interacción entre
las regiones citosolicas de las proteínas integrales de las dos membranas.
Las proteínas clave que
participan en estas interacciones se conocen como SNARE.
Fusión
entre las membranas de la vesícula y el blanco.
Cuando las vesículas
artificiales de lípidos (liposomas) que contiene SNARE-t purificada se mezclan
con liposomas que contienen SNARE-v purificada, los dos tipos de vesículas se
fusionan entre sí pero no las vesículas del mismo tipo.
Este hallazgo indica que las
interacciones entre proteínas SNARE-t y v son capaces de unir dos bicapas de
lípidos con la fuerza suficiente por sí misma para inducir la fusión dentro de
la célula.
Vesículas Cubiertas con cop
ll TRANSPORTE DE CARGAMENTO DEL RETICULO ENDOPLASMICO AL APARATO DE GOLGI
Transporte de cargamento del
retículo endoplasmatico al aparato de Golgi
Las vesículas cubiertas con
COPII median la primera rama del traslado por la vía biosintética, del RE al
ERGIC y la red cis de Golgi. Está cubierta contiene varias proteínas que se
identificaron en las células de las levaduras.
La cubierta COP II
selecciona y concentra ciertos componentes para transportar en vesículas.
Ciertas proteínas integrales de membrana del RE se capturan en forma selectiva
porque contienen señales de exportación del RE como parte de su cola
citosolicas.
Las proteínas seleccionadas
por las vesículas cubiertas con COP II incluyen:
1) enzimas que actúan en las
etapas avanzadas de la vía biosintética, como las glucosiltransferasas del
aparato de Golgi
2) proteínas de membrana
participantes en el acoplamiento y fusión de la vesícula con el compartimiento
blanco
3) proteínas de membrana que
pueden unirse con cargamento soluble.
Entre estas proteínas se
encuentra una pequeña proteína G llamada Sar 1, que tiene función reguladora,
en el inicio de la formación de la vesícula y la regulación del ensamblaje de
la cubierta de la vesícula.
1) Sar 1 es reclutada en la
membrana del RE en la forma unida a GDP y es inducida a intercambiar su GDP por
un GTP por una proteína llamada GEF (factor de intercambio de guanina) (al
unirse al GTP, Sar1 sufre un cambio que hace que la hélice alfa en su extremo N
terminal se inserte en la hoja citosolicas de la bicapa del RE
2) Este evento induce la
curvatura de la bicapa lipídica en ese sitio. (Constituye una formación de una
membrana aplanada a una esférica)
3) Sar1-GTP atrajo dos poli
péptidos adicionales a la cubierta COP II, Sec23 y Sec24, que se unen con forma
de banana, (Sec 24 funciona como proteína adaptadora de la cubierta COPII
4) Las subunidades restantes
de la cubierta COPII Sec23 y Sec 32, se unen con la membrana para formar la
jaula estructural externa de la cubierta proinica.
Una
vez que se ensambla toda la cubierta COPII la yema se separa de la membrana del
RE en la forma de una vesícula cubierta por COP II. Antes que la vesícula
cubierta pueda fusionarse con una membrana blanco, la cubierta proteínica debe
desensamblarse y sus componentes se liberan al citosol.
Vesículas
cubiertas con CLATRINA.
Las vesículas de estas
cubiertas contienen:
Una celosía externa parecida
a un panal formada por la proteína clatrina, la cual constituye un soporte
estructural.
Una capa interna formada por
adaptadores de proteína que cubre la superficie de la membrana de la vesícula y
que está dirigida hacia el citosol.
Formación
de vesículas cubiertas con clatrina en la red trans de Golgi.


Coroideremia
Este padecimiento se
caracteriza por presentar invariablemente degeneración retiniana y de coroides.
Está determinada
genéticamente, con un modo de herencia recesivo ligado al cromosoma X, por lo
que la mayoría de estos pacientes son masculinos.
El primer signo de
afectación ocular consiste en ceguera nocturna que inicia en la infancia
temprana que progresivamente conduce a la ceguera total alrededor de los 20 años
de edad, aunque algunos pierden la visión en forma total hasta los 45 años.
Síndrome
de Lowe (síndrome oculocerebrorrenal).
El síndrome de Lowe u
oculocerebrorrenal es un trastorno genético que se hereda en forma recesiva
ligada al cromosoma X.
Se caracteriza por cataratas
congénitas, disfunción renal tubular y déficit neurológico.
El desarrollo de las
cataratas comienza desde la 7a. a 9a. semana de gestación, debido a anomalías
en la migración del epitelio del cristalino.
Durante el periodo neonatal
inmediato los pacientes presentan proteinuria, aminoaciduria, fosfaturia y
acidosis metabólica, así como retardo mental, hipotonía, trastornos de la
conducta y ausencia de reflejos osteotendinosos profundos.
Enfermedad de Menkes
En el síndrome de Menkes,
las células en el cuerpo pueden absorber el cobre, pero son incapaces de
liberarlo.
Es causado por un defecto en
el gen ATP7A.
Cobre se puede acumular en
el intestino delgado y los riñones, pero los niveles bajos de este elemento en
otras zonas pueden afectar la estructura de huesos, piel, cabello y vasos
sanguíneos e interferir con la función nerviosa.
Es hereditario, lo cual
significa que se transmite de padres a hijos.
El gen está en el cromosoma
X.
Mulcolipidosis
II
Es una enfermedad que se
caracteriza por la presencia de múltiples inclusiones en el citoplasma de los
fibroblastos.
Se debe a que existe una
señalización anómala de enzimas lisosoma es en las células del tejido
mesenquimatoso, como las hidrolasas lisosomales durante su paso por Cis-golgi
adquieren residuos de manosa 6-fosfato.
·
Se presenta a muy temprana edad
·
Retraso mental progresivo
·
Malformaciones esqueléticas
·
Los pacientes mueren en la primera década de
vida.
Mulcolipidosis III
Tambien llamada
Polidistrofia Pseudo-Hurler, son entidades monogenicas y con un modo de
herecencia autosomico recesivo.
Se debe a que existe una
señalización anómala de enzimas lisosoma es en las células del tejido
mesenquimatoso, como las hidrolasas lisosomales durante su paso por Cis-golgi
adquieren residuos de manosa 6-fosfato.
·
Se presenta a muy temprana edad
·
Retraso mental progresivo
·
Malformaciones esqueléticas
·
Anomalias cardiovasculares
Presenta una progresion mas
lenta en el deterioro mental, viven hasta la edad adulta.
Deficiencia
de glicoproteínas
Presenta alteraciones en a
las glicoproteinas sericas (exportacion) que
consisten en un peso molecular menor al esperado debido a un bajo nivel
de glucosilacion.
Proteina de union a la
tiroxina
Factores del complemento c3a
y c4a
·
Presentan retraso psicomotor
·
Alteraciones dermatologicas
·
Oftalmologicas
·
Y disfuncion hepatica


ENDOCITOSIS
Y EXOCITOSIS
Endocitosis:permite el flujo
de entrada de tipo pinocitosis(Ingestión de pequeñas
partículas),fagocitosis(Ingestión de grandes partículas) , por receptor
(Captación de macromoléculas extracelulares).
Exocitosis: Permite el flujo
de salida..
Endocitosis
Definición: Es
un proceso en el cuál la célula interioriza los receptores de la superficie
celular y ligando extracelulares unidos a ellos:
Fagocitosis, endocitosis por
volumen y Endocitosis mediada por un receptor
Plegamiento de membrana que
forma vesículas.
• Necesita
consumo de ATP (hidrólisis de ATP).
• Reposición
de la membrana.
Una vez en el interior de la
célula, las vesículas de endocitosis pueden seguir dos caminos
1Digestion: Las
vesículas de endocitosis se fusionan con lisosomas primarios para formar
vacuolas digestivas. Los productos de la digestión se incorporarán después al
metabolismo celular.
2Transito intracelular: Algunas
vesículas de endocitosis simplemente transportan su contenido desde un punto de
la célula a otro.
En las células que revisten
los vasos sanguíneos se transportan sustancias desde la sangre hasta tejidos
periféricos.
- Participa
en la ingestión, secuestro y degradación
de sustancias captadas del espacio extracelular
- Es
el proceso en el cual la célula ingiere macromoléculas, material
particulado, y otras sustancias desde el espacio extracelular
- El
material de endocitosis se va a englobar en una vesícula, si esta es
pequeña se llamara pinocitosis y la vesícula será pinositotica.
- Si
la vesícula será grande el método se llamara fagocitosis y la vesícula es
un fagosoma
- Si
la vesícula es pequeña, el tipo de endocitosis se conoce como
pinocitosis(célula que bebe) y la vesícula es una vesícula pinositotica
- Se
divide en fagocitosis y pinocitosis
FAGOCITOSIS: Proceso
de englobamiento de material partículas grande, lo llevaran a cabo los
fagocitos.
Engloba: Microorganismos,
fragmentos celulares y células.
• El
material que se ingiere es muy grande.
• La
célula extiende unas prolongaciones de membrana llamada pseudópodos, que rodean progresivamente a la partícula hasta
formar un fagosoma.
• Estos
materiales se digieren en los lisosomas.
• Fagosoma:
vesícula de gran tamaño.
ENDOCITOSIS POR VOLUMEN(PINOCITOSIS: Es
la ingestión de pequeñas partículas o líquidos, mediante la formación de
vesículas muy pequeñas , se da en todo tipo de células.
Se da Enterocitos, túbulos
proximales renales, epitelio del epidídimo y en los linfocitos. Es muy rápida
en los macrófagos.
Determina procesos:
• Incorporación
de ciertas sustancias (lipoproteínas).
• Destrucción
de receptores de superficie
• Entrada
de virus en las infecciones
- En
todos esos actúan los lisosomas que degradan esas moléculas
- Una
vez que el contenido de la vesícula ha sido procesado, la membrana de la
vesícula vuelve a la superficie de la célula.
- Puede funcionar como el reciclaje de membrana entre
la superficie celular y los compartimentos interiores
• ENDOCITOSIS
MEDIADA POR UN RECEPTOR : Captación de
macromoléculas extracelulares especificas (ligandos),después de su unión con
receptores en la superficie externa de la membrana plasmática
• Proporciona un medio para la captación de muchos tipos
diferentes de ligandos como: hormonas/ factores de crecimiento/enzimas/ proteínas
sanguíneas que transportan ciertos nutrientes.
• ENDOCITOSIS POR RECEPTORES DE MEMBRANA. : Se
trata de sustancias que primero deben acoplarse a moléculas receptoras
específicas, los receptores se encuentran agrupados en la membrana y están
unidos n la parte citosólica con proteínas clatrinas, o se agrupan después de
haberse unido a las moléculas que serán transportadas.
• Las
células humanas incorporan el colesterol el cual es utilizado en la síntesis de
membranas y también como precursor de otros esteroides.


Exocitosis
Proceso por el cual una vesícula se mueve desde el
citoplasma hacia la membrana plasmática, desde donde pone su contenido en el
espacio extracelular
Las
vesículas secretoras se forman en vesículas en la red tras-Golgi
Por
Exocitosis (se van a fusionar con la membrana)
Inicia
en el retículo endoplasmico
1) Mecanismo
por secreción constitutiva
•
Las sustancias se exportaran en vesículas de transporte enviadas
a la membrana plasmática
•
Las proteínas que abandonan las células por
este proceso se secretan después de una síntesis y salen del aparato de Golgi
q ej.
Secreción de inmunoglobulinasà
plasmocitos
•
Presente en todas las células
2) Mecanismo
por secreción regulada
Células
especializadas ej. Células endocrinas/exocrinas/neuronas concentran las
proteínas de secreción y las almacenan temporalmente en vesículas secretoras
dentro del citoplasma.
Para
que ocurra la secreción tiene que activarse un fenómeno regulador (estimulo
hormonal o nervioso)
Sucede
en la liberación de los gránulos de cimógeno por las células principales de la
mucosa gástrica
El
estímulo señal causa la entrada temporal Ca+ en el citoplasma
Estimula
las vesículas de secreción para que se fusione con la membrana plasmática
Libera
su contenido hacia el exterior
Anexo:
•
No se acumula producto de secreción
•
Pocas vesículas se ven en el citoplasma
LISOSOMAS

Características generales
Son organelos digestivos
ricos en enzimas hidrolíticas como proteasas, nucleasas, glucosidasas, lipasas
y fosfolipasas.
Representan un compartimento
digestivo principal en la célula que degrada macromoléculas derivadas de los
mecanismos endocíticos, así como de la célula misma en un proceso conocido
como autofagia.
Las enzimas de un lisosoma
comparten una propiedad importante: todas alcanzan su actividad óptima en un pH
ácido, por lo que son hidrolasas acidas.
El pH óptimo de estas
enzimas se sitúa por debajo del pH del
compartimiento lisosómico, que se aproxima a 4.6.
Aunque tienen una colección
predecible de enzimas, su apariencia en las micrografías electrónicas no es
distintiva ni uniforme.
Función
·
Degradación de materiales que llegan a la
célula desde el ambiente extracelular.
Muchos organismos
unicelulares ingieren partículas de alimento que luego degradan en un lisosoma.
Los nutrientes obtenidos pasan por la membrana lisosómica hacia el citosol.
En los mamíferos, las
células fagocíticas funcionan como eliminadores que ingieren los detritos y
microorganismos que pudieran ser peligrosos
Las bacterias ingeridas casi
siempre se desactivan por el pH bajo del lisosoma y luego se someten a la
digestión enzimática.
Membrana lisosómica
Tiene una estructura
fosfolipídica inusual que contiene colesterol y un lípido exclusivo denominado
acido lisobifosfatídico.
La mayor parte de las
proteínas estructurales de la membrana lisosómica se clasifican en:
·
Proteínas de membrana asociadas con lisosomas
(LAMP)
·
Glucoproteínas de membrana lisosómica (LGP)
·
Proteínas integrales de membrana lisosómica
(LIMP).
Las LAMP, LGP y LIMP
representan más del 50 % del total de las proteínas de la membrana lisosómica
y están muy glucosiladas en la superficie luminal.
Transportan productos
finales de la digestión (aminoácidos, sacáridos, nucleótidos) hacia el
citoplasma, donde se utilizan en los procesos sintéticos de la célula o sufren
exocitosis.
Una ATP-asa de H+ tipo V:
Transportan iones H+ a la luz lisosómica, manteniendo un pH ácido.
Clasificación funcional
Lisosomas primarios
Son vesículas que se forman a partir del aparato de Golgi, contienen únicamente hidrolasas ácidas y tienen la propiedad de fusionarse con diversos tipos de vesículas fagocíticas.
Leucocitos
·
Neutrófilos
·
Eosinófilos
·
Monocitos
Lisosomas secundarios
Contienen tanto las
hidrolasas ácidas como los materiales que van a ser degradados , Son lisosomas
primarios fusionados con otras sustancias, de origen interno o externo.
Se distinguen en dos tipos
·
Vacuolas heterofágicas
·
Vacuolas autofágicas
Vesículas autofágicas
·
Se piensa que las vesículas autofágicas se
forman a partir de membranas del RE que contienen fosfatasa ácida.
·
Estas membranas envuelven a aquellas
partículas que van a ser degradadas.
·
Inicialmente estas vesículas (autofagosomas)
no contienen enzimas lisosomales; a ellas se les fusionan lisosomas, con lo
cual se inicia la degradación del contenido de las vesículas autofágicas; estas
nuevas vesículas se conocen como autofagolisosomas.
·
Se pueden identificar fácilmente con el
microscopio electrónico, porque en su interior se pueden observar mitocondrias,
retículo endoplásmico, glucógeno y otros tipos de entidades citoplásmicas con
varios grados de desorganización.
Fagolisosomas
·
Se les conoce como heterofágicas, para
distinguirlas de las autofágicas.
·
Se encuentran en células fagocíticas
profesionales, como: monocitos, macrófagos, células de Kupffer, etc.
·
Contienen, además de las enzimas lisosomales,
partículas ajenas a la célula que han sido capturadas por un proceso de
fagocitosis.
·
La formación del fagolisosoma es un mecanismo
de defensa que el organismo posee para eliminar virus, bacterias, parásitos
unicelulares, células cancerígenas, partículas microscópicas, etc., que
penetran en él por diferentes vías.
Endosomas
Pueden considerarse como
organulos citoplasmaticos estables o estructuras transitorias formadas como
resultado de endocitosis .
Estas vesículas se
distinguen por:
·
Su morfología
·
Ser menos densas que los lisosomas
·
Por ser
un compartimento ligeramente ácido (pH 6 a 6.2).
La pinocitosis es un
mecanismo muy importante que la célula posee para transportar a su interior
sustancias solubles extracelulares.
Endosomas tempranos
·
Suelen encontrarse en el citoplasma màs
periferico
·
Tiene una estructura tubulovesicular
·
Poseen un medio apenas màs acido (PH 6,2 a
6,5)
·
Clasifican y reciclan proteinas
interiorizadas por vias endociticas.
Endosomas tardíos
·
Suelen posicionarse cerca del aparato de
Golgi y del núcleo.
·
Posee una estructura más compleja y con
frecuencia exhiben membranas internas.
·
Su PH es más acido, con un promedio de 5,5.
Cuerpos multivesiculares
(MVB)
·
Son transportadores muy selectivos
·
Se distinguen fácilmente, son vacuolas en las que en su lumen se pueden
distinguir vesículas más pequeñas.
·
Debido a que su pH es ligeramente ácido (pH
5.5 a 6) y contienen hidrolasas ácidas, son considerados como lisosomas
secundarios.
Cuerpos residuales
·
En el lumen de este tipo de vesículas se
observa una estructura polimórfica electrodensa compuesta por residuos no
digeridos por los lisosomas.
·
Algunos cuerpos residuales son negativos para
la tinción de fosfatasa ácida y para otras enzimas lisosomales, por lo cual se
les considera como poslisosomas.
·
En el
hígado, la secreción ocurre hacia los conductos biliares, o hacia los
túbulos en el riñón.
·
Los cuerpos residuales suelen denominarse
pigmento de degaste .
·
Son una caracteristica normal del
envejecimiento celular.
·
La ausencia de ciertas enzimas lisosomicas
puede causar la acumulaciòn de sustrato no digerido en los cuerpos residuales.
·
Transtornos graves que en forma colectiva se
denominan Enfermedades por deposito lisosomal
Acidificación de lisosomas y
endosomas
·
Es una proteína integral de membrana,
compuesta por lo menos por nueve subunidades, que en su conjunto tienen un
tamaño de 750 kDa.
·
El espacio interior de endosomas, lisosomas y
fagolisosomas se mantiene a un pH más ácido que el pH del citosol o del espacio
extracelular.
·
Es mediada por una bomba de protones
electrogénica, llamada bomba vacuolar de protones, o ATPasa tipo V para H+.
·
Transporta en contra de gradiente protones
presentes en citosol.
·
La concentración de protones en lisosoma es
al menos cien veces mayor que en el citosol.
Autofagia
·
Principal mecanismo celular por el cual
varias proteínas citoplasmáticas, orgánulos o estructuras celulares son
degradadas en los lisosomas.
·
Mantiene un equilibrio bien controlado entre
las funciones celulares anabólicas y catabólicas
·
Permite que la célula elimine los orgánulos
innecesarios o no deseados.
·
En general la autofagia puede dividirse en
tres mecanismos:
Macroautofagia:
·
Proceso no especifico en el cual una porción
del citoplasma o un orgánulo completo es rodeado por una membrana doble
(membrana de aislamiento o fagóforo).
·
Para formar una vesícula secuestrante
denominada Autofagosoma.
·
Una vez que autofagosoma se completa, se
fusiona con un lisosoma y genera autofagolisosoma, en el cual el organelo
encerrado se degrada y los productos de degradación se hacen disponibles para
la célula.
Microautofagia
·
Es un proceso no especifico en el cual las
proteínas citoplasmáticas son degradadas en un proceso lento y continuo en
condiciones fisiológicas normales.
·
Las proteínas citoplasmáticas solubles
pequeñas se incorporan dentro de los lisosomas por invaginación de la membrana
lisosómica.
Autofagia
mediada por chaperonas
·
Es el único proceso selectivo de degradación
proteica y requiere la colaboración de chaperonas citosólicas especificas como
la proteína chaperona de choque térmico denominada hsc73.
·
Es responsable de la degradación de
aproximadamente el 30% de las proteínas citoplasmáticas en órganos como el
hígado y el riñón.
·
Se activa durante la privación de sustancias
nutritivas y necesita la presencia de señales de localización en las proteínas
que se han de degradar y de un receptor especifico en la membrana lisosómica.
·
La hsc73 se fija a la proteína y contribuye a
su transporte a través de la membrana lisosómica hacia la luz, donde
finalmente se degrada.
Enzimas lisosomales
·
Se sintetizan en el RER y se clasifican en el aparato de Golgi
·
Las enzimas lisosomales son glicoproteínas
que contienen cadenas glicosídicas unidas a residuos de serina (O-glicosídicas)
o de asparagina (N-glicosídicas).
·
Son sintetizadas en ribosomas adheridos a las
membranas del retículo endoplásmico rugos
Enfermedades:
Enfermedades por almacenamiento lisosómico
v Son un grupo de trastornos hereditarios, que
producen sus primeros síntomas generalmente en la niñez o adolescencia,
acortando la expectativa de vida y provocando grados variables de discapacidad
en las personas afectadas.
v Están causadas por una alteración genética y mal
funcionamiento de las enzimas lisosomales, lo cual provoca la acumulación de
diferentes sustancias no metabolizadas en el lisosoma con alteración
de la función y muerte celular.
Leucodistrofia metacromatica
Leucodistrofia metacromática:
lipidosis sulfátida): la enzima faltante es la
arilsulfatasa A.El nombre se refiere a disfunción de sustancia blanca cerebral
metacromasia
Gaucher
Existe una deficiencia en la enzima Glucocerebrosidasa y la
principal sustancia almacenada es el glucocerebrosido.
TAY-SACHS
Existe un deficiencia en la enzima hexosaminidasa β y la principal sustancia acumulada es el Gangliosido Gm2
Existe una deficiencia en la
degradacion lisosomica de los gangliosidos que se encuentra dentro de las
estructuras laminillares concentricas
Sandhoff
Deficiencia enzimática: Hexosaminidasas
A y B
Producto acumulado: Gangliósido
GM2 y globósido
Manifestación clínica:
Idéntica a Tay-Sachs.
Defecto enzimático de Hexosaminidasa
A: los gangliósidos no pueden degradarse, se acumulan en
lisosoma formando corpúsculos que contienen colesterol y
fosfolípidos, acaban lesionando gravemente las neuronas.
Defecto enzimático de Hexosaminidasa
B: interfiere en la degradación de glicolípidos con el mismo azúcar
terminal que los gangliósidos (globósido), se acumulan en riñones, hígado
y bazo.
Krabbe
Deficiencia enzimática: Galactocerebrosidasa
Producto acumulado: Galactocerebrósido
La acumulación afecta al
crecimiento de la vaina protectora de mielina del nervio y provoca una gran degeneración
de las habilidades motoras.
 Niemann-Pick
Niemann-Pick
Deficiencia enzimática: Esfingomielinasa
Producto acumulado: Esfingomielina
Manifestación clínica:
Retraso mental, crecimiento del hígado y bazo, hinchazón abdominal, mancha rojo
fresa en parte de atrás del ojo, dificultades de alimentación.
Depósito origina una
alteración en las células deteriorándolas, deformándolas y terminando en muerte
de estas.
Gangliosidosis GM1
Existe una deficiencia de la enzima: Galactosidasa β GM1
FABRY
Fabry(lipidosis ceramida
trihexosido): La enzima faltante es la α-galactosidasa y su principal sustancia
almacenada es el globotriosilceramida.Esta ligada al cromosoma X
Aspartilglucosaminuria
Deficiencia enzimática: N-aspartil-beta-glucosaminidasa
Producto acumulado: Oligosacáridos
N-ligados
Manifestación clínica:
Afectación del esqueleto, retraso del desarrollo psicomotor,
hepatomegalia, opacidades del cristalino, trastornos del comportamiento,
estatura corta.
-Manosidosis
Deficiencia enzimática: α-Manosidasa
Producto acumulado: α-Manósidos
Manifestación clínica:
Pie equino, inmunodeficiencia, anomalías esqueléticas, discapacidad auditiva,
trastorno progresivo de las funciones mentales y del habla, ataxia.
Este defecto produce una
acumulación de manosilglucoproteínas, fundamentalmente en el sistema nervioso
central y en el hígado, junto con una excreción aumentada de
manosiloligosacáridos en orina.
Síndrome de Hurler
Deficiencia enzimática: a-L-iduronidasa
Producto acumulado: Dermatán
sulfato, heparán sulfato
Manifestación clínica:
Estatura baja, engrosamiento progresivo de los rasgos faciales,
miocardiopatías, pérdida auditiva, retraso mental y en el desarrollo.
Resulta en la acumulación de
grandes cantidades de glicosaminoglicanos en las células del tejido conectivo,
incluido cartílago y hueso.
Sindrome de hurter(MPS ll)
Este es un transtorno por degradacìòn de los glucosaminoglicanos
Existe una insuficiencia proteica de L-idurato sulfatasa y el producto
acumulado es el Dermatàn sulfato ,heparan sulfato.
Sindrome de Maroteaux-Lamy (MPS
lV)
Transtorno por degradaciòn de los glucosaminoglicanos
Existe una deficiencia proteica de GalNAc 4-sulfatasa/arilsulfatasa y el
producto acumulado es el Dermatan sulfato.
Pompe
Existe una deficiencia
proteica de α-1,4-Glucosidasa
Producto acumulado:Glucogeno
Wolman
Deficiencia enzimática: Lipasa
ácida
Producto acumulado: Ésteres
de colesterol, TAG
Manifestación clínica:
Vómitos, diarrea, esteatorrea, hepatomegalia, esplenomegalia, distensión
abdominal, deterioro capacidades mentales, anemia grave, caquexia.
La deficiencia de esta
enzima conduce a un almacenamiento masivo en las células, de triglicéridos y
ésteres de colesterol. Estos depósitos dañan los tejidos afectados,
principalmente el cerebro, hígado y bazo.
Canavan
Deficiencia enzimática: Aspartoacilasa
Producto acumulado: Ácido
N-acetilaspártico
Manifestación clínica: Se
distinguen dos formas:
*Canavan grave presentan:
Hipotonía grave, retraso en el desarrollo, otras alteraciones neurológicas,
concentración alta de ácido n-acetil-L-aspártico en orina, sangre y LCR.
*Canavan leve presenta:
leve retraso en el desarrollo, problemas al hablar , NAA en orina ligeramente
elevado.
Ocasiona la acumulación
del ácido N-acetilaspártico lo cual interfiere en el crecimiento de las
vainas de mielina de las neuronas, que son las que permiten una
transmisión eficiente del impulso nervioso.
Danon
 Deficiencia enzimática: LAMP2 (Proteína de Membrana Asociada Lisosómica 2)
Deficiencia enzimática: LAMP2 (Proteína de Membrana Asociada Lisosómica 2)
Producto acumulado: Presencia de vacuolas autofágicas
Manifestación clínica: Miocardiopatía severa, debilidad muscular
esquelética variable, retraso mental, palpitaciones, dolor en el pecho,
problemas para respirar, calambres.
Las
células sin la LAMP2, proteínas de fusión entre vacuolas autofágicas y lisosomas
ocurre más lentamente, lo cual puede llevar a la acumulación de vacuolas
autofágicas.
Cistinosis

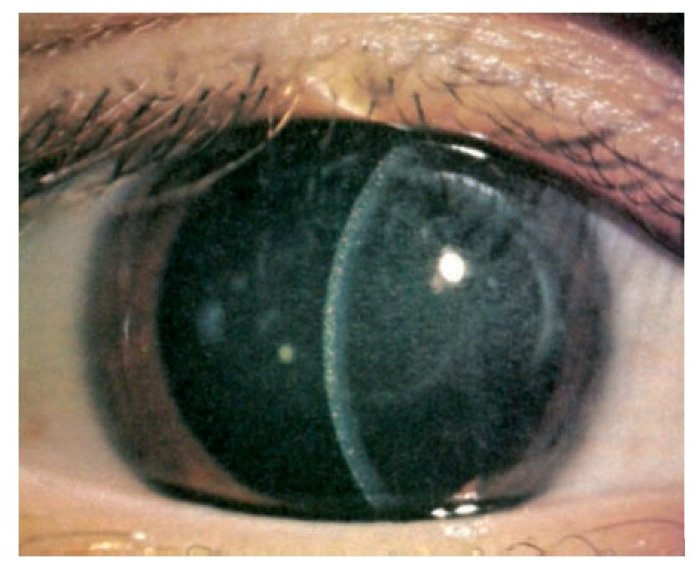
Deficiencia proteica: Cistinosina (transportador de cistina)
Producto acumulado: cistina
El
defecto en la sintesis de el transportador lisosomico de cistina se caracteriza
por la acumulacion de cistina libre en los lisosomas.
Mucolipidosis ll (enfermedad
de celulas de inclusión )
Deficiencia proteica: GlcNAc-1-fosfotransferasa(conduce al envio
defectuoso de las enzimas lisosomicas hidroliticas más solubles ).
Gota
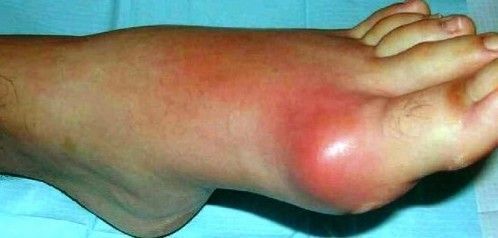 El
ácido úrico proveniente del catabolismo de las purinas se produce en exceso,
provoca la deposición de cristales de urato en las articulaciones. Los
cristales son fagocitados por las células y se acumulan en los lisosomas
secundarios; estos cristales provocan la rotura de dichas vacuolas con la
consiguiente liberación de enzimas lisosómicas en el citosol, causa la
digestión de componentes celulares, la liberación de sustancias de la célula y
la autolisis celular.
El
ácido úrico proveniente del catabolismo de las purinas se produce en exceso,
provoca la deposición de cristales de urato en las articulaciones. Los
cristales son fagocitados por las células y se acumulan en los lisosomas
secundarios; estos cristales provocan la rotura de dichas vacuolas con la
consiguiente liberación de enzimas lisosómicas en el citosol, causa la
digestión de componentes celulares, la liberación de sustancias de la célula y
la autolisis celular.
Síntomas:
v Articulaciones del dedo gordo del pie, rodilla o
tobillo
v El dolor comienza súbitamente, durante la noche. Se
describe como pulsátil, opresivo o insoportable.
v Articulación luce caliente y roja, está muy sensible
e hinchada
v Puede haber fiebre.
Artritis reumatoide
Causa
la destrucción de las membranas lisosomales, con la consecuente liberación de
las enzimas y la lisis celular.
Síntomas:
v La rigidez matutina, que dura por más de 1 hora, es
común.
v Dolor articular
v Dolor torácico al respirar
v Resequedad en ojos y boca

Tratamiento
El
único tratamiento específico, seguro y eficaz disponible hoy en día lo
constituyen las Terapias de Reemplazo Enzimático
Las
proteínas recombinantes, aplicadas por vía intravenosa cada una o dos semanas,
reemplazan las enzimas faltantes o deficientes en los pacientes.
PEROXISOMAS
“Son vesículas simples limitadas por membranas, que pueden contener un
centro denso y cristalino de enzimas oxidativas”.
Orgánulos rodeados de membrana con muchas oxidasas, que generan peróxido de
hidrógeno
Se encargan de la desintoxicación de la célula, particularmente de la
degradación de los ácidos grasos de cadena muy larga.
Se encuentran en las células eucariotas.

Localizado cerca del RE
Papel esencial en el metabolismo lipídico
“ORGANELOS MULTIFUNCIONALES”
+ de 50 enzimas que participan (β- oxidación) de cadena muy larga (
24-26 carbonos).
Síntesis de plasmalógenos localizados en la mielina,
colesterol y ácidos biliares.
Son orgánulos celulares que desempeñan una función primordial
en la utilización del oxígeno. Morfológicamente se parecen a los lisosomas,
pues son partículas esféricas, limitadas por una membrana, de un diámetro
variable que oscila entre 0.3 y 1.5 µm, y con un contenido enzimático
Las enzimas que tienen no son hidrolasas ácidas, sino
enzimas que intervienen en el metabolismo del H2O2 que junto con las
mitocondrias y cloroplastos colaboran en ciertas funciones.
Se observaron por primera vez en el riñón y en el hígado
de roedores (1950), y se les denominó microcuerpos.
Después se fueron
localizando en muchos tipos celulares de vertebrados, protozoos, levaduras y en
algunos tipos celulares de vegetales.
Con el tiempo, se observan peroxisomas en casi todos los
tipos celulares, al punto que ya se consideran un orgánulo habitual de las
células.
Se encargan de la desintoxicación de la célula, en
especial de la degradación de los ácidos grasos de cadena muy larga.
Se denominan así porque sus enzimas utilizan el “O2
molecular” para eliminar átomos de “Hidrogeno” de sustratos específicos, por
medio de una reacción oxidativa que produce H2O2.
El H2O2 es un metabolito muy tóxico, por lo cual la
enzima catalasa se encarga de
degradarlo a oxígeno y agua
CATALASA
40% de la proteína total de los peroxisomas del hígado
Catabolismo de purinas
1) Los ácidos nucleicos viejos se degradan primero en nucleótidos y después
en bases púricas y pirimidinas.
2) Estas bases son reutilizan para la síntesis de nuevos ácidos nucleicos o
degradadas.
3) En la degradación de las bases púricas intervienen diversas enzimas del
peroxisoma. El H2O2 que se libera con estas oxidaciones es descompuesto por la
catalasa.
4) El acido úrico se degrada a alantoína, para después ser degradado a acido
glioxilico y urea y finalmente ser convertida en amoniaco.
Los peroxisomas de las células animales intervienen en el metabolismo de
los lípidos realizando procesos que tienen lugar en otros orgánulos.
Entre un 10 y un 25% de los ácidos grasos se degradan en peroxisomas y el
resto, en mitocondrias y este proceso de degradación se denomina β-oxidación y conduce a la formación de
acetil-CoA.
En células animales (el colesterol, el dolicol y los ácidos biliares)
pueden ser sintetizados en los peroxisomas, además de en el REL.
METABOLISMO DEL ÁCIDO GLICÓLICO
El ácido glicólico es un subproducto de la fotosíntesis de los
cloroplastos, producido por la fijación de O2 en la enzima RUBISCO.
El ácido glicólico entra en los peroxisomas y es oxidado a ácido
glioxílico, el cual se convierte en glicina, que pasa de los peroxisomas a las
mitocondrias donde se transforma en serina y CO2.
CONVERSIÓN de GRASAS-HIDRATOS DE CARBONO: CICLO DEL GLIOXILATO
Tiene lugar en algunos órganos vegetales, y se ha estudiado con
detenimiento en la semilla de ricino, y cuando se produce la germinación de
esta semilla, la grasa del endosperma se convierte en glúcidos
Las moléculas de Acetil-coa que se producen en la degradación de ácidos grasos
se usan para producir acido succínico, en el proceso ciclo del glioxilato.
El ácido succínico produjo en este ciclo, abandona los glioxisomas y
penetra en las mitocondrias, cuya matriz
es oxidado en C-Krebs a ácido oxalacético, que abandona las mitocondrias y se
convierte en glucosa en el hialoplasma.
SINDROME DE ZELLWEGER
Su origen etiológico se encuentra en una anomalía
genética que da lugar a una producción deficiente del peroxisoma, que causan la
muerte en la lactancia temprana.
Caracteriza
por una acumulación anormal de ácido fitánico, de colesterol o ácidos biliares,
en diversas áreas como el cerebro, hígado o riñones.
Se presenta dimorfismo craneofacial, anormalidades del
esqueleto, extremidades proximales cortas, encefalopatía, convulsiones,
anormalidades oculares como retinopatía, cataratas y enfermedad del hígado. (reconocible por alteraciones neurológicas,
visuales y hepáticas)
Las personas con esta enfermedad carecen de peroxisomas
en las células hepáticas y renales, y aun no existe alguna cura para esta
enfermedad.
ADRENOLEUCODISTROFIA
Conocida como Adrenomieloneuropatía o Suprarrenoleuco
Distrofia (ADL), es causada por un defecto
de una proteína de membrana que transporta ácidos grasos de cadena muy larga
(VLCFA) hacia los peroxisomas, con la
estructura del peroxisoma intacta, ligada al cromosoma X.
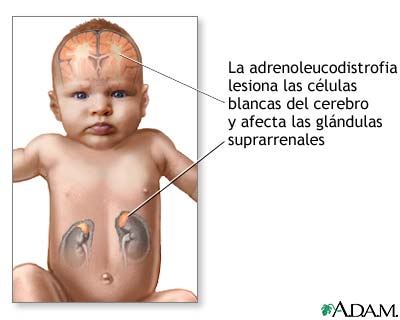 Existe una falla en la β- oxidación
de los ácidos grasos, almacenamiento anormal de lípidos en el cerebro, medula y
glándulas suprarrenales.
Existe una falla en la β- oxidación
de los ácidos grasos, almacenamiento anormal de lípidos en el cerebro, medula y
glándulas suprarrenales.
Se
produce la acumulación de (VLCFA) en los testículos, hígado, medula
suprarrenal, cerebro.
Los
niños no presentan síntomas hasta la parte media de la infancia.
Enfermedad de refsum infantil
Es
una enfermedad metabólica neurodegenerativo de peroxisomas, perteneciente al
grupo de las lipidosis.
Es
hereditaria y se transmite de padres a hijos según un patrón autosómico
recesivo.
Se
parecida al síndrome de Zellweger, pero es distinta en adultos.
Se
manifiesta por la retinitis pigmentosa, nistagmo, ataxia cerebelosa, sordera de
percepción, deformidades esqueléticas e ictiosis, hepatomegalia, así como
neuropatía periférica.
• La
esperanza de vida puede llegar a los 20 años.
Concentraciones elevadas de acido fitánico, acido
pristanico, acido pipecolico, ácidos ditrihidroxicolestanoico y AGCML
Formación
de nuevos peroxisomas en una célula se puede producir de dos formas: a) por
crecimiento y división de los preexistentes, y b) por generación a partir del
retículo endoplasmatico.
a)El
crecimiento y proliferación de los peroxisomas también puede darse por la
participación del retículo endoplasmático.
En concreto, desde las cisternas del retículo endoplasmático se pueden formar
por evaginación y escisión estructuras membranosas de tipo vesicular con todas
las moléculas típicas de los peroxisomas que por fusión irán creando peroxisomas
maduros. Pero incluso, una vez formado el peroxisoma, las proteínas que
formarán parte de la membrana, y algunas internas, además de desde el citosol,
pueden llegar en vesículas producidas en el retículo, por una ruta vesicular
independiente de COPII.

MITOCONDRIA
La estructura de la
mitocondria se puede observar en el microscopio electrónico como organelos
individuales de estructura arriñonada con longitud que va de 1 a 4 um. Un
aspecto muy importante de estos organelos es que tienen la posibilidad de
fusionarse o bien fisionarse (es decir, dividirse en dos).
Estos organelos son conocidos por su función más característica que es la generación de moléculas de ATP que se utilizan en actividades celulares que requieren energía. Si bien la función primordial de la mitocondria es la producción de energía, cuenta con otras características funcionales como por ejemplo: la producción de ciertos aminoácidos o bien ña generación de grupos hemo, también tiene función importante en la captación y liberación de iones calcio.
Estos organelos son conocidos por su función más característica que es la generación de moléculas de ATP que se utilizan en actividades celulares que requieren energía. Si bien la función primordial de la mitocondria es la producción de energía, cuenta con otras características funcionales como por ejemplo: la producción de ciertos aminoácidos o bien ña generación de grupos hemo, también tiene función importante en la captación y liberación de iones calcio.
La estructura y composición
básica de esta unidad funcional es que cuenta con dos membranas mitocondriales
(interna y externa) y una matriz mitocondrial.
 La membrana mitocondrial
externa rodea por completo a la mitocondria mientras que la memebrana interna
se subbdivide en dos dominios interconectados contando con proteínas residentes
que tienen distintas funciones, esta membrana en su dominio mas predominante
(el dominio con relación a la matriz mitocondrial) es la única que envuelve a
las crestas mitocondriales. Estas crestas contienen una gran cantidad de la
maquinaria productora de ATP denominada ATP sintasa.
La membrana mitocondrial
externa rodea por completo a la mitocondria mientras que la memebrana interna
se subbdivide en dos dominios interconectados contando con proteínas residentes
que tienen distintas funciones, esta membrana en su dominio mas predominante
(el dominio con relación a la matriz mitocondrial) es la única que envuelve a
las crestas mitocondriales. Estas crestas contienen una gran cantidad de la
maquinaria productora de ATP denominada ATP sintasa.
Las membranas mitocondriales
dividen al organelo en dos compartimientos acuosos, uno al interior denominado
matriz mitocondrial y el otro entre las dos membranas llamado espacio
intermembrana.
La membrana externa esta
formada por lípidos y enzimas que
participan en la oxidación de adrenalina, la degradación de triptófano y la
elongación de los AG. En cambio la membrana interna contiene alrededor de 100
polipeptidos diferentes, esta es rica en cardiolipina (difosfatidilglicerol)
cuya función es importante para facilitar la actividad de las proteínas
participantes en la síntesis de ATP.
La matriz tienen una consistencia gelatinosa por
elevada concentración de proteínas hidrosolubles. Además de tener proteínas
también contiene ribosomas que son un poco más pequeños que los que encontramos
en le citosol, también encontramos moléculas de ADN a las que denominaremos
ADNmitocondrial, con estos datos podemos decir que la mitocondria tiene su
propio material genético con la capacidad de producir su propio ARN y por ende
producir sus propias proteínas.
METABOLISMO OXIDATIVO
Tenemos que comentar
primeramente el metabolismo de la glucosa los llevan a cabo las enzimas de la
glucolisis que se encuentran en el citosol, este proceso se lleva a cabo por
diez reacciones enzimáticas que ya las hemos estudiado. Pero durante este
proceso podemos decir que se lleva a cabo la producción de dos moléculas de ATP
por solo la oxidación de una molécula de glucosa, producción de dos NADH, también produce dos moléculas de piruvato que
es la molécula de la que preside el metabolismo oxidativo en la mitocondria.
Los organismos aerobios son capaces de extraer grandes cantidades de energías del piruvato y NADH producidos durante la glucolisis, estos producirán más de 30 moléculas de ATP. Cada molécula de piruvato se trasporta hacia la matriz mitocondrial donde se descarboxila para formar acetil-coA esta reacción se da por la acción de un complejo multienzimatico denominado piruvato deshidrogenasa.
Los organismos aerobios son capaces de extraer grandes cantidades de energías del piruvato y NADH producidos durante la glucolisis, estos producirán más de 30 moléculas de ATP. Cada molécula de piruvato se trasporta hacia la matriz mitocondrial donde se descarboxila para formar acetil-coA esta reacción se da por la acción de un complejo multienzimatico denominado piruvato deshidrogenasa.
Ciclo de Krebs
 La acetil-coA ingresa a una
vía denominada ciclo de los ácidos
tricarboxilicos en la que se oxida el sustrato y se conserva su energía,
todas las enzimas del ciclo se encuentran en la matriz mitocondrial excepto
una, la succinato deshidrogenasa que se encuentra en la membrana mitocondrial
interna y tiene una estrecha relación en la cadena transportadora de
electrones.
La acetil-coA ingresa a una
vía denominada ciclo de los ácidos
tricarboxilicos en la que se oxida el sustrato y se conserva su energía,
todas las enzimas del ciclo se encuentran en la matriz mitocondrial excepto
una, la succinato deshidrogenasa que se encuentra en la membrana mitocondrial
interna y tiene una estrecha relación en la cadena transportadora de
electrones. En esta vía metabólica central por excelencia ya que ahí convergen las demás vías metabólicas ya sea catabólicas o anabólicas. Este ciclo está dado por 8 reacciones cuya finalidad es transformar energía y dar un sustrato siguiente como lo es el oxalacetato para que vuelva a iniciar el ciclo. La producción neta del ciclo son 3 NADH, 1 FADH y 1 GTP por molécula de acetil-coA.
Importancia
de las coenzimas reducidas en la formación de ATP
Podemos decir que los
productos primarios del ciclo son las coenzimas reducidas NADH y FADH, estas
contienen electrones de alta energía removidos de varios sustratos durante su
oxidación. Estas coenzimas son de vital importancia en las reacciones aeróbicas
y de igual manera contribuyen a la producción de ATP.
Los electrones de estas
moléculas pasan a una serie de proteínas que se encuentran en la membrana
mitocondrial interna, esta serie de proteínas se le denomina cadena
transportadora de electrones, dichas partículas pasan a lo largo de la cadena
respiratoria en reacciones que liberan energía. La energía liberada durante el
transporte de electrones se almacena en forma de un gradiente de concentración
de protones a través de la membrana. Los electrones de baja energía se
transfieren al receptor final que es el oxígeno (O2).
Una enzima proporciona la
energía necesaria para fosforilar moléculas de ADP y convertirlas en ATP esta
enzima recibe el nombre de ATP-asa, esta formación es gracias al gradiente
electroquímico de protones que se formó durante la cadena transportadora de
electrones.
Transporte
de electrones
Los electrones de alta
energía unidos al FADH y NADH se transfieren a través de una serie de
acarreadores específicos de electrones que constituyen a la cadena
trasportadora de electrones. Esta se compone de 5 de portadores de electrones
unidos a la membrana: flavoproteinas, citocromo, átomos de Cu, ubiquinona y
proteínas de hierro y azufre.
1. Las flavoproteinas
consisten en un polipeptido unido con fuerza a un grupo prostético relacionado;
FAD (FMN. Cada uno es capaz de aceptar y donar dos electrones y dos protones.
2. Citocromos,
proteínas que cuentan con grupos prostéticos hemo, el átomo de Fe da lugar a la
aceptación y perdida de un electrón. Hay tres tipos distintos de citocromos
(a,b,c).
3. 3
átomos de Cu, se localizan dentro de un solo complejo. Aceptan y donan un solo
electrón.
4. Ubiquinona,
molécula liposoluble que contiene una cadena hidrófoba larga. Cada ubiquinona
puede aceptar y donar dos electrones y dos protones.
Complejos
Complejo 1. NADH
deshidrogenasa, es la puerta de entrada de la cadena y cataliza la
transferencia de un par de electrones del NADH a lla ubiquinona.
Complejo 2. Succinato deshidrogenasa, esta formado por
4 polipeptidos, dos subunidades hidrófobas que fijan la proteína a la membrana
y dos subunidades hidrofilicas que comprende a la enzima del ciclo de Krebs.
Suministra una vía para alimentar al FAD y a la ubiquinona. Contiene un grupo
hemo el cual atrae electrones que se han escapado, lo que evita que se formen
radicales superoxidos destructivos.
Complejo 3. Citocromo bc1, cataliza la transferencia
de electrones de la ubiquinona al citocromo c, se forman 4 protones por cada par
de electrones que se transfieren a este complejo.
Complejo 4. Citocromo c
oxidasa, es el paso final del transporte de electrones en la mitocondria, es la
transferencia sucesiva de dichas partículas del citocromo c reducido al
oxígeno. Es aquí donde se lleva a cabo la verdadera respiración celular.

ATP-asa
Es un complejo proteico en
forma de hongo conformado por dos componentes principales, una cabeza F1
esférica y una sección basal F0 incrustada en la membrana interna. Ahora bien
gracias al potencial gradiente electroquímico que se creo durante la cadena
transportadora de electrones, se dice que cuando entran 4 protones en la parte
basal o F0 de la enzima, le da la fuerza necesaria llamada fuerza protón motriz
que gracias a esta se lleva a cabo la fosforilacion que tiene como interacción
al ADP y Pi para la formación de ATP.
Mecanismo
para la formación de ATP.
1. La energía liberada con el movimiento del
protón no se usa para impulsar la fosforilación del ADP en forma directa, sino
que se emplea en cambiar la afinidad de unión del sitio activo por el producto
ATP.
2.
Cada sitio activo progresa de manera sucesiva
por tres diferentes conformaciones con afinidades distintas por los sustratos y
los productos.
3.
El ATP se sintetiza por catálisis rotatoria,
en la que una parte de la ATP sintasa .
rota en relación con otra parte.
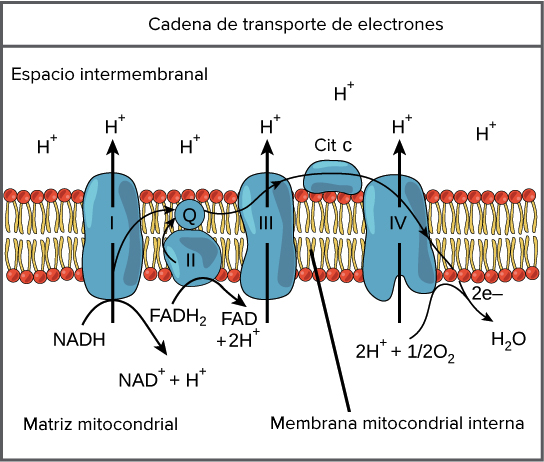
rota en relación con otra parte.
Otras
funciones para la fuerza motriz de
protones además de la síntesis de ATP.
Aunque la producción de ATP
puede ser la actividad más importante de las mitocondrias, estos organelos
participan en muchos otros procesos que requieren el ingreso de energía. A
diferencia de la mayor parte de los organelos que dependen sobre todo de la
hidrólisis del ATP para impulsar sus actividades, las mitocondrias dependen de
una fuente alternativa de energía: la fuerza motriz de protones. Por ejemplo,
esta fuerza motriz de protones impulsa la captación de ADP y Pi en las
mitocondrias a cambio del ATP y H+, respectivamente.
Esta y otras actividades que ocurren durante la respiración aeróbica. En otros ejemplos, la fuerza motriz de los protones puede usarse como fuente energética para “jalar” iones calcio hacia el interior de la mitocondria; para impulsar los fenómenos de la fusión mitocondrial, y para hacer que polipéptidos específicos seleccionados entren a la mitocondria desde la matriz.
Esta y otras actividades que ocurren durante la respiración aeróbica. En otros ejemplos, la fuerza motriz de los protones puede usarse como fuente energética para “jalar” iones calcio hacia el interior de la mitocondria; para impulsar los fenómenos de la fusión mitocondrial, y para hacer que polipéptidos específicos seleccionados entren a la mitocondria desde la matriz.
El modo en que los niveles
de ATP tienen una función importante en el control de la velocidad de la
glucólisis y el ciclo del ácido tricarboxílico mediante la regulación de la
actividad de enzimas clave. Dentro de la mitocondria, el nivel de ADP es el
factor determinante de la velocidad respiratoria.
Cuando los niveles de ADP son bajos, los de ATP casi siempre son altos, por lo que no hay necesidad de oxidar más sustrato para proporcionar electrones a la cadena respiratoria.
En tales condiciones, la síntesis ATP es escasa, por lo que los protones no pueden reingresar a la matriz mitocondrial a través de la sintasa de ATP. Esto conduce a la acumulación sostenida de fuerza motriz de protones, lo que a su vez inhibe las reacciones de bombeo de protones en la cadena de transporte de electrones y el consumo de oxígeno por la citocromo oxidasa.
Cuando la proporción ATP/ADP disminuye, el consumo de oxígeno aumenta en forma súbita. Ya se demostró que muchos factores influyen en la velocidad de la respiración mitocondrial, pero las vías que regulan la respiración no se conocen bien.
Cuando los niveles de ADP son bajos, los de ATP casi siempre son altos, por lo que no hay necesidad de oxidar más sustrato para proporcionar electrones a la cadena respiratoria.
En tales condiciones, la síntesis ATP es escasa, por lo que los protones no pueden reingresar a la matriz mitocondrial a través de la sintasa de ATP. Esto conduce a la acumulación sostenida de fuerza motriz de protones, lo que a su vez inhibe las reacciones de bombeo de protones en la cadena de transporte de electrones y el consumo de oxígeno por la citocromo oxidasa.
Cuando la proporción ATP/ADP disminuye, el consumo de oxígeno aumenta en forma súbita. Ya se demostró que muchos factores influyen en la velocidad de la respiración mitocondrial, pero las vías que regulan la respiración no se conocen bien.
Función
anormal de las mitocondrias.
La mayor parte de las
enfermedades causadas por una función anormal de las mitocondrias se
caracteriza por degeneración del tejido muscular o cerebral, ya que los dos
utilizan cantidades enormes de ATP.
La gravedad de estos trastornos varía desde enfermedades que causan la muerte durante la lactancia, hasta los que causan convulsiones, ceguera, sordera y/o episodios parecidos a un accidente vascular cerebral, y hasta trastornos leves caracterizados por intolerancia al ejercicio o espermatozoides inmóviles. Los pacientes con alteraciones graves casi siempre tienen fibras musculares esqueléticas anormales que contienen grandes agregados periféricos de mitocondrias.
La gravedad de estos trastornos varía desde enfermedades que causan la muerte durante la lactancia, hasta los que causan convulsiones, ceguera, sordera y/o episodios parecidos a un accidente vascular cerebral, y hasta trastornos leves caracterizados por intolerancia al ejercicio o espermatozoides inmóviles. Los pacientes con alteraciones graves casi siempre tienen fibras musculares esqueléticas anormales que contienen grandes agregados periféricos de mitocondrias.
Más de 95% de los polipéptidos
de la cadena respiratoria son codificados por genes que residen en el núcleo. Y
sin embargo, la mayoría de las mutaciones vinculadas con enfermedades
mitocondriales se han rastreado hasta mutaciones en el DNA mitocondrial, o
mtDNA. Los trastornos más graves casi siempre se originan de mutaciones (o
deleciones) que afectan genes que codifican RNA de transferencia mitocondrial,
el cual es necesario para la síntesis de los 13 polipéptidos producidos en las
mitocondrias humanas.
La herencia de los
trastornos mitocondriales contrasta en varias formas con la herencia mendeliana
de los genes nucleares.
Las mitocondrias presentes en las células de un embrión humano provienen exclusivamente de las mitocondrias que estaban en el óvulo al momento de la concepción, sin contribución alguna del espermatozoide que lo fecundó. Por consiguiente, los trastornos mitocondriales se heredan por línea materna.
Las mitocondrias presentes en las células de un embrión humano provienen exclusivamente de las mitocondrias que estaban en el óvulo al momento de la concepción, sin contribución alguna del espermatozoide que lo fecundó. Por consiguiente, los trastornos mitocondriales se heredan por línea materna.
Además, es posible que las
mitocondrias de una célula contengan una mezcla de mtDNA normal (tipo nativo) y
mutante, un trastorno conocido como heteroplasmia. Es muy probable que se
acumulen mutaciones mitocondriales en las células que permanecen mucho tiempo
en el cuerpo, como las del tejido nervioso y muscular. Se cree que los cambios
degenerativos en la función mitocondrial contribuyen a varias enfermedades
neurológicas frecuentes de inicio en el adulto, en particular la enfermedad de
Parkinson (PD).

ENFERMEDADES

LHON está
causada por mutaciones en el ADN mitocondrial (ADNmt). Todas producen
alteraciones en los genes MT-ND1, MT-ND4 y MT-ND6 del
complejo I de la cadena respiratoria mitocondrial.
Entre los 18 y
los 30 años se observa con frecuencia una pérdida de visión central aguda o
subaguda, brusca e indolora.
Afecta a ambos
ojos simultáneamente o de forma secuencial, con pérdida de visión en el segundo
ojo semanas o meses después.
También pueden
darse otros signos neurológicos. Estas anomalías se conocen como Leber
"plus", e incluyen trastornos motores, distonía, temblor postural y
ataxia cerebelosa.
PEARSON
Dicho síndrome
es entonces un disturbio mitocondrial congénito, de carácter raro, que se
da normalmente como consecuencia de un rearreglo del ADN mitocondrial o también
pero menos frecuentemente por causa de mutaciones.
El síndrome de
Pearson se caracteriza por una anemia sideroblástica refractaria, vacuolización
de los precursores de la médula ósea y disfunción exocrina del páncreas.
BARTH
El BTHS está
causado por mutaciones en el gen TAZ (tafazzin; Xq28) que
codifica la aciltransferasa Taz1p implicada en el metabolismo de la
cardiolipina, un fosfolípido muy importante de las membranas mitocondriales
internas. Lo cual compromete finalmente la estructura mitocondrial o la función
de la cadena respiratoria.
•
Miocardiopatia
•
Neutropenia
•
Músculos esqueléticos subdesarrollados y
debilidad muscular
•
Crecimiento retrasado
•
Intolerancia al ejercicio
•
Deficiencia de cardiolipina
•
Aciduria
BERI-BERI
DEFICIENCIA DE
VITAMINA B1:
•
Consumo de alcohol
•
Genético
•
Las mujeres embarazadas y las personas con
hipertiroidismo
•
La diarrea prolongada
•
Los niños que se alimentan con leche materna o
leche maternizada con bajo contenido de vitamina B1.
•
La diálisis renal
•
Existen dos tipos de esta enfermedad: beriberi
húmedo y beriberi seco. El beriberi húmedo puede afectar la función cardíaca y,
en los casos más extremos, puede ocasionar insuficiencia cardíaca. El beriberi
seco provoca daños en los nervios y puede ocasionar pérdida de la fuerza
muscular y, eventualmente, parálisis muscular. Si no la enfermedad no se
detecta ni se trata, puede ocasionar la muerte.
•
Deficiencia del complejo 3:
•
Encefalomiopatía mortal infantil, acidosis
láctica congénita, hipotonia, postura distrófica, convulsiones y coma. Es común
la fibrosis roja.
•
Encefalomiopatía de brotes posteriores: estatura
corta, ataxia, demencia, pérdida auditiva, neuropatía sensorial, retinopatía
pigmentosa y síntomas piramidales. Es común la fibrosis roja. Posibilidad de
acidosis láctica.
•
Miopatía, con intolerancia al ejercicio que
deviene en debilidad continua. Es común la fibrosis roja. Posibilidad de
acidosis láctica.
•
Cardiomiopatía
.
•
Deficiencia del complejo 4:
•
Encefalomiopatía: normal durante los primeros 6
a 12 meses de vida; después presenta regresión en el desarrollo, ataxia,
acidosis láctica, atrofia óptica, oftalmoplegia, nistagmus, distonia, signos
piramidales y problemas respiratorios. Frecuentes convulsiones .
•
Miopatía: dos variantes principales:
•
Miopatía infantil mortal
•
Miopatía infantil benigna
Síndrome de
Leigh
Síntomas son
la falta de succión en los bebés y la pérdida de control de la cabeza y de los
movimientos voluntarios. pérdida de apetito, vómitos, irritabilidad, llanto
continuo y convulsiones. Si la enfermedad progresa, los
síntomas incluyen debilidad generalizada, falta de tono
muscular, espasticidad, trastornos del movimiento, incapacidad para
coordinar el equilibrio
El síndrome de Leigh se puede clasificar en:
ADN
mitocondrial o Síndrome de Leigh de transmisión (o herencia) materna
ADN
nuclear: causada por una mutación en el ADN nuclear, y sub-dividido de
acuerdo al modo de herencia y del gen mutado en la forma con
herencia autosómica recesiva y la forma con herencia ligada al
cromosoma X.
Síndrome de
Kearns-Sayre
Sintomas: sordera
bilateral neurosensorial, las afectaciones cardiacas, las afectaciones del
sistema nervioso central, la miopatía del músculo esquelético, los desórdenes
intestinales y endocrinos y el fallo renal.
El KSS está
causado por deleciones de grandes fragmentos de ADN mitocondrial (ADNmt),
resultando en la pérdida de genes involucrados en la ruta de señalización de la
fosforilación oxidativa.
Tratamiento:
colocación de un marcapasos ,proporcionar audífonos. El suplemento con coenzima
Q10 ha resultado ser beneficioso en algunos casos. Las manifestaciones
oftalmológicas se pueden tratar quirúrgicamente.
MELAS
Síntomas:
cefaleas con vómitos y, en ocasiones, signos de pseudoapoplejía, como confusión,
hemiparesia y hemianopsia.
A menudo se
producen en pacientes con síntomas crónicos como debilidad muscular, sordera,
diabetes, baja estatura, miocardiopatía, retraso en el desarrollo, pérdidas de
memoria o trastornos de atención.
Causada por:
mutaciones en el ADN mitocondrial (3243A> G en el gen del ARNt de leucina)
WOLFRAM
Síntomas:
diabetes mellitus tipo I, diabetes insípida, atrofia óptica y signos
neurológicos, atonia del tracto urinario, ataxia, neuropatía periférica,
demencia, problemas psiquiátricos y/o epilepsia
Hiperamonemia
1 y 2
HIPERAMONEMIA
I: Es el defecto en la falta de la enzima carbamoil fosfato sintetasa I, la
cual transforma el amoniaco a carbamoilfosfato.
Síntomas:
- NH4 elevado en:
- Sangre
- Hígado
- Orina
- Vómitos
- Retraso mental
- NH4 elevado en:
- Sangre
- Hígado
- Orina
- Vómitos
- Retraso mental
HIPERAMONEMIA
II: Es el defecto en la falta de la enzima ornitina transcarbamilasa, la cual
transforma el carbamoilfosfato a citrulina. Es una enfermedad de herencia
ligada al cromosoma X.
Síntomas:
- Glutamina elevada en:
- Sangre
- LCR
- Glutamina elevada en:
- Sangre
- LCR
- Vómitos, convulsiones, retraso mental
- Letal en varones en periodo neonatal
- Letal en varones en periodo neonatal
Encefalopatía
mitocondrial
Herencia: La
mutación 3271T> C del gen ARNt Leu
v
Síntomas :
cefaleas con
vómitos y, en ocasiones, signos de pseudoapoplejía, como confusión, hemiparesia
y hemianopsia.
A menudo se
producen en pacientes con síntomas crónicos como debilidad muscular, sordera,
diabetes, baja estatura, miocardiopatía.
No existe
ningún tratamiento que hasta hoy haya sido exitoso.
Comentarios
Publicar un comentario